Ambertools 安装
- 系统 Linux
- 安装
conda install -c conda-forge ambertools=22 compilers***只需要Ambertools即可,这里选择安装ambertools22. ***
蛋白文件准备
- 由于蛋白氨基酸含有非标准氨基酸,所以加氢时,可以考虑使用MOE或者Chimera等软件进行加氢处理。
- 加氢结束以后,将非标准氨基酸残基命名为OLS,提取非标准氨基酸后,使用pdb4amber对蛋白删除氢原子并重新编号,保存为Protein.pdb
非标准氨基酸需要合理的氢原子位置,所以先进行上述操作。
如果不是Ambertools22最后能识别的氢,整个蛋白的氢需要删除,同时,也要注意氨基酸残基名字的修改,当然可以优先使用H++ Server,但需要先删去非标准氨基酸上的多余原子最后再补加或者替换。
非标准氨基酸准备
以氨基酸接一个糖元为例子,改非标准氨基酸如下图: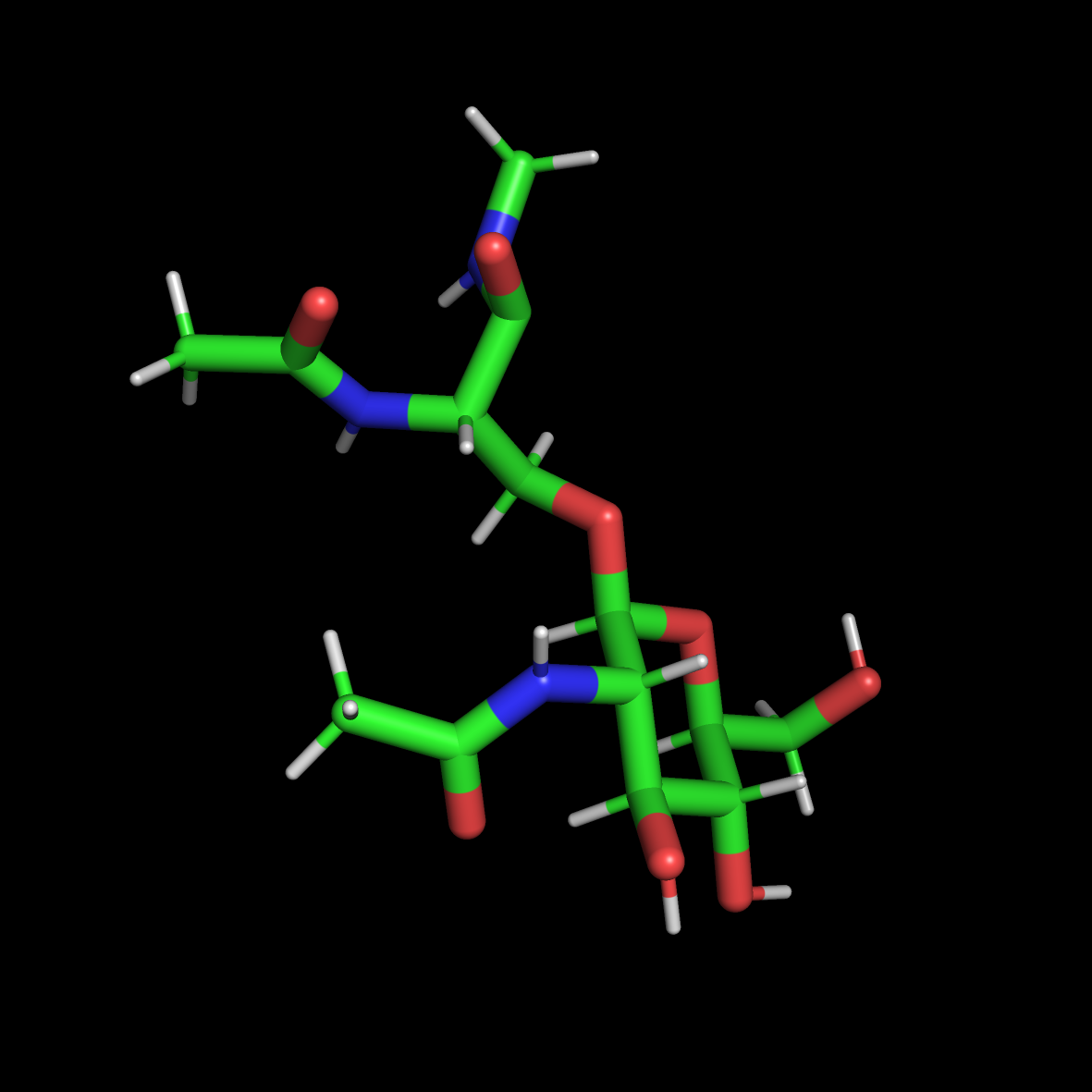
主要做了如下处理:
- 从蛋白提取该非标准氨基酸
- 对其进行了ACE,NMe封端
- 保存成OLSME.pdb文件
- 对所得OLSME.pdb文件进行修改,确保各个原子的编号的唯一性,不存在多余的TER等标识,残基名修改成OLS
***可以不进行ACE, NMe处理,只进行甲基化,或者更简单的加氢处理,这样得到的pdb文件更单一,不会存在太多问题。
非标准氨基酸参数的生成
使用bcc电荷
- antechamber -fi pdb -i OLSME.pdb -bk OLS -fo ac -o OLSME.ac -c bcc -at amber
- 制作mc文件,保存为OLSME.mc。最好有GaussView程序

- HEAD_NAME 与前一个残基连接的原子的原子名称
- TAIL_NAME 与后一个氨基连接的原子的原子名称
- MAIN_CHAIN 主链原子的名称
- OMIT_NAME 残基不存在的原子名称,即补氢,补甲基,补ACE, NMe的原子都要写
- RRE_HEAD_TYPE
POST_TAIL_TYPE 前后连接的原子的原子类型- CHARGE 残基总电荷
- prepgen -i OLSME.ac -o OLSME.prepin -m OLSME.mc -rn OLS,会产生一个NEWPDB.PDB文件,可以通过pymol查看该文件,可知mc文件是否书写正确
- parmchk2 -i OLSME.prepin -f prepi -o OLSME_parm10.frcmod -a Y -p $AMBERHOME/dat/leap/parm/parm10.dat
检查生成OLSME_parm10.frcmod文件,如有”ATTN, need revision”标识则需要用gaff进行补充
- grep -v “ATTN” OLSME_parm10.frcmod > parm10_OLSME.frcmod, 如有上述情况,则需要进行这个操作,如果没有则不需要
- parmchk2 -i OLSME.prepin -f prepi -o gaff_OLSME.frcmod
使用RESP电荷
#p B3LYP/6-311G** opt Pop=MK IOp(6/33=2,6/42=6) scrf使用高斯16进行计算- bcc电荷方案的第一步操作改为 antechamber -fi gout -i OLSME.log -bk OLS -fo ac -o OLSME_GAU.ac -c resp -at amber
- 将上述bcc电荷方案所得的OLSME.ac文件中的坐标和电荷替换成OLSME_GAU.ac里面的坐标及文件,并保存为OLSME.ac文件中的坐标和电荷替换成OLSME_GAU
- 余下步骤同bcc电荷方案
生成prmtop, inpcrd文件
tleap.in:
source leaprc.protein.ff14SB
source leaprc.water.tip3p
set default PBRadii mbondi3
loadamberprep OLSME.prepin
loadamberparams OLS_gaff.frcmod
loadamberparams parm10_OLS.frcmod
mol = loadpdb Protein.pdb
savepdb mol Protein_H.pdb
saveamberparm mol Protein_dry.prmtop Protein_dry.inpcrd
solvatebox mol TIP3PBOX 12.0
addions mol Na+ 0
addions mol Cl- 0
savepdb mol Protein_solv.pdb
saveamberparm mol Protein_solv.prmtop Protein_solv.inpcrd
quit
运行tleap -s -f tleap.in
结果: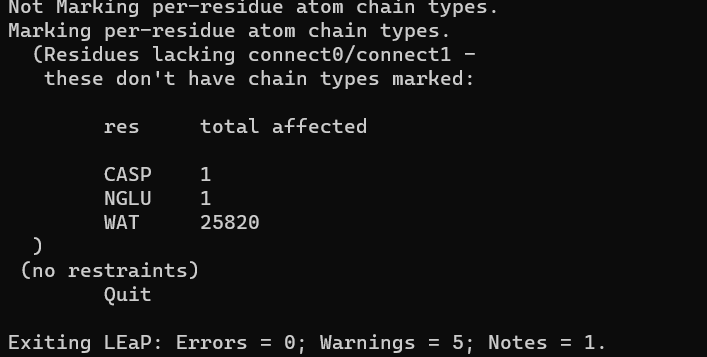
查看生成的Protein_H.pdb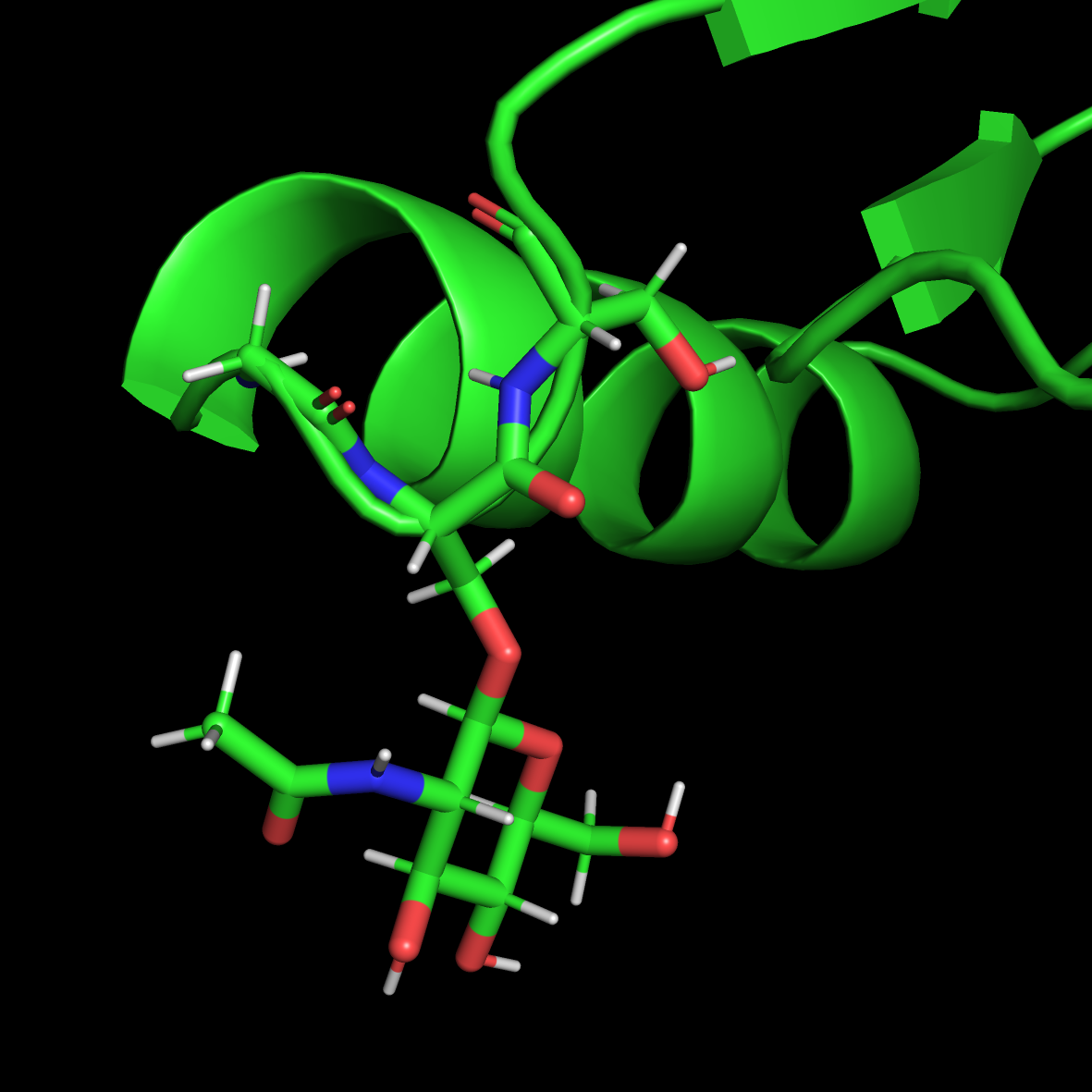
生成GROMAC运行文件
acpype -p Protein_solv.prmtop -x Protein_solv.inpcrd -d
生成一个protein_solv.amb2gmx文件,查看Protein_solv_GMX.gro文件。
注:如有问题,请发邮件hanlan8702@163.com